INTRODUCTION
Globally, the burden of disease associated with Streptococcus pneumoniae (pneumococcus) infections is considerable, a situation which is compounded by the fact that treatment of pneumococcal infection has become increasingly challenging due to the growing threat of antibiotic resistance1. Cigarette smoking, still prevalent in many countries, is recognized as being one of the most prominent independent risk factors for invasive pneumococcal disease2. Moreover, current smokers who develop pneumococcal community-acquired pneumonia (CAP) have a striking 5-fold increase in the risk of 30-day mortality irrespective of age, co-morbidities and early implementation of guideline-concordant antimicrobial chemotherapy3.
Tobacco-induced susceptibility for development and severity of pneumococcal infection is attributed to impairment of pulmonary host defences, favoring colonization and invasion of the upper and lower respiratory tract, respectively4. With the exception of our previous studies5,6, relatively little attention has, however, focused on the direct effects of smoking on the pathogen per se. These studies were the first to document that exposure of the pneumococcus to cigarette smoke condensate (CSC) in vitro results in genesis of biofilm5,6. Biofilm is a highly-hydrated viscoelastic extracellular matrix, comprised of various types of bacterium-derived polymeric materials, which not only insulates the pathogen against host defences but also obstructs penetration of antibiotics.
Resistance of the pneumococcus to macrolide antibiotics represents a problem of increasing magnitude as these agents are widely used in the treatment of pneumococcal infection7. As opposed to mutations in genes that confer antibiotic resistance, resistance of the pneumococcus to macrolides results from the horizontal transfer of two types of intact resistance genes. These are, firstly the gene encoding ribosomal methylase, which obstructs target binding, and secondly those encoding drug efflux pumps7.
Building on our previous studies on mechanisms of smoking-related antibiotic resistance and pneumococcal disease, the current investigation was undertaken to determine whether exposure to cigarette smoke condensate (CSC) of two strains of S. pneumoniae, harboring distinct genetic mechanisms of macrolide resistance, affected the expression of these antibiotic resistance genes. These were strains 2507 and 521 of the pathogen, both serotype 23F, commonly linked to invasive disease. The former expresses the erm(B) ribosomal methylase gene and the latter the macrolide efflux pump gene, respectively.
METHODS
CSC from Murty Pharmaceuticals (Lexington, KY, USA) was dissolved to 40 g/L in dimethylsulfoxide (DMSO). The total amount of condensate8 generated during the combustion of a single cigarette is 26.3 mg, considerably higher than the concentrations of CSC used in the current study. Briefly, the two strains of the pneumococcus were grown overnight to mid-log growth phase in tryptone soy broth (TSB, Merck, Darmstadt, Germany) and optically standardized to 2.0×108 colony-forming units (cfu)/ mL. The bacteria were then exposed to CSC (two successive 90 min exposures at final concentrations of 80 or 160 mg/L), followed by the addition of TSB or the macrolide antibiotic, clarithromycin (2 and 8 mg/L for strains 521 and 2507, respectively), for 15 min at 37oC, 5% CO2. These concentrations of CSC were based on our previous studies on pneumococcal biofilm formation5,6, while those of clarithromycin were based on minimum inhibitory concentration (MIC) values for strains 521 and 2507 of 2 and >256 mg/L, respectively9. DMSO solvent controls were included in all experiments. The bacterial cells were then concentrated by centrifugation, the pellet snap frozen in liquid nitrogen and stored at -80oC prior to undergoing a two-way RNA extraction process as described previously6.
Following RNA extraction, a high-capacity complementary DNA (cDNA) reverse transcription kit (Applied Biosystems, Foster City, CA, USA) was used according to the manufacturer’s instructions for the generation of cDNA.
Clarithromycin resistance gene expression was determined using real-time reverse transcription polymerase chain reaction (qRT-PCR) using Stratagene Brilliant II SYBR® Green QPCR low ROX master mix (Agilent, Santa Clara, CA, USA) and PIKOREAL 96 well plates (ThermoScientific Inc.) on a PIKOREAL 96 real-time detection system (ThermoScientific Inc.), as described previously6. In the case of erm(B), the forward primer used was 5’– AGGGCATTTAACGACGAAAC–3’, while the reverse primer was 5’–GACGCATGGCTTTCAAAAAC–3’. The forward and reverse primers used for the antimicrobial resistance gene mef(A) were 5’–CTTTTCATACCCCAGCACTC–3’ and 5’– GCAATCACAGCACCCAATAC–3’, respectively. The housekeeping genes, gyr(A) and gyr(B) were also included.
Relative gene expression was performed10 by comparing the relative change in expression of the target genes to that of the reference genes and normalized to the untreated strains to reflect the log(10-ΔΔCq). The results are expressed as mean ± standard deviation (SD) of the log-fold increase of three different experiments with duplicate measurements for each system. Analysis of variance was measured using repeated-measures ANOVA with Tukey-Kramer multiple comparisons test as post-examination. Statistical significance was set at p<0.05.
RESULTS
Expression of the erm(B) and mef(A) genes by strains 2507 and 521 of the pneumococcus, respectively, was undetectable in the absence of either CSC or clarithromycin. On the other hand, as shown in Figure 1A, exposure of strain 2507 of the pneumococcus to CSC alone, in the absence of clarithromycin, caused dose-related induction of the erm(B) macrolide resistance gene, which achieved statistical significance (p<0.05) at a concentration of 160 mg/L of the condensate. In addition, and as expected, exposure of strain 2507 of the pneumococcus to clarithromycin alone resulted in significant expression of the erm(B) gene, which was further and significantly (p<0.001) augmented by prior exposure of the pathogen to CSC at a concentration of 160 mg/L. The magnitude of gene expression for the system treated with the combination of clarithromycin and CSC at 160 mg/L was 27% higher (p<0.05) than that of the sum of the systems treated with the antibiotic and CSC individually, consistent with an augmentative interaction. Although not statistically significant, the corresponding increase for the system treated with the combination of clarithromycin and CSC at 80 mg/L was 25%.
Figure 1
The effects of clarithromycin (Clari, 8 mg/L) and CSC (80 and 160 mg/L), alone and in combination, on the expression of erm(B) by strain 2507 (Figure 1A) and mef(A) by strain 521 (Figure 1B) of S. pneumoniae.
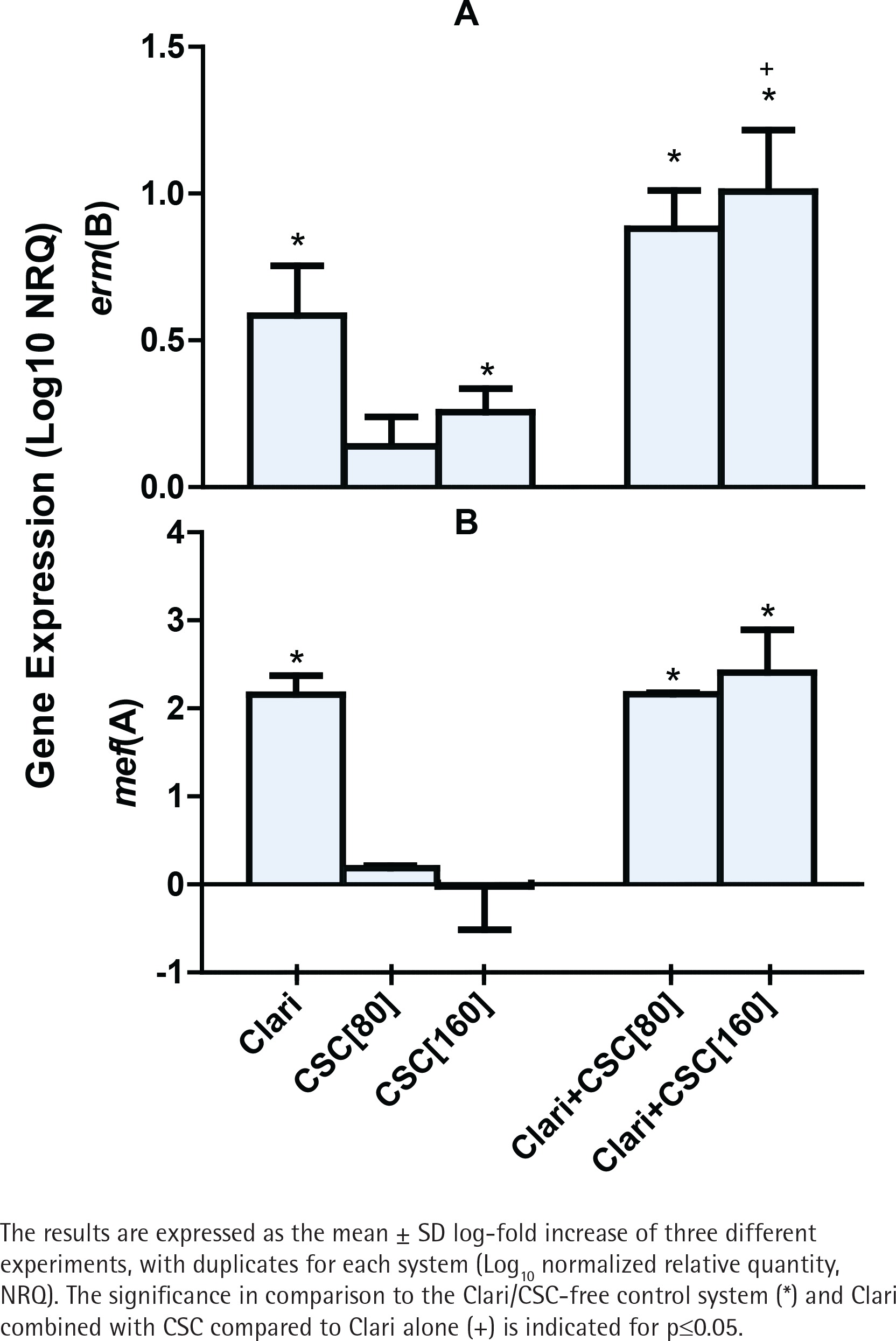
In the case of strain 521, as shown in Figure 1B, expression of the mef(A) gene was significantly increased in the presence of clarithromycin alone, the magnitude of which was unaffected by prior exposure of the pathogen to CSC, while exposure to CSC alone had no detectable effect on gene expression. These differential effects of CSC on expression of the erm(B) and mef(A) genes may result from lack of effect of CSC on the transcriptional mechanisms involved in the induction of the mef(A) gene.
DISCUSSION
Gene Expression (Log10 NRQ) erm(B)
Our findings imply that smoking sensitizes macrolideresistant pneumococci in the airways for increased expression of the erm(B) gene. This proposed scenario is based on two observations. Firstly, that exposure of strain 2507 of the pneumococcus to CSC alone resulted in spontaneous induction of the erm(B) gene, a remarkable finding, which, to our knowledge has not been described previously. Secondly, that the magnitude of expression of the erm(B) gene following sequential exposure of the pneumococcus to CSC and clarithromycin was significantly greater than the sum of the individual exposures to CSC and the antibiotic.
These effects of CSC on both spontaneous induction and augmentation of clarithromycinmediated expression of the erm(B) gene may predispose to the development of a more aggressive resistance phenotype, characterized by a more rapid onset and greater magnitude of antibiotic resistance. This contention is based on a previous study, which demonstrated that following macrolide-mediated induction of the erm(B) gene, which happens rapidly, completion of the post-translational events is slow, with acquisition of the fully-resistant phenotype, only occurring after a lengthy lag period of up to twelve hours11,12. In the context of the findings of the current study, this lag period may be significantly shortened due to prior smoke-mediated induction of erm(B).
CSC-mediated induction of the erm(B) gene, is likely to be the consequence of a general stress response of the pneumococcus to CSC-mediated oxidative stress. In this context, cigarette smoke contains an abundance of pro-oxidative toxicants, including organic and inorganic highly-reactive free radicals and heavy metals, which trigger, directly or indirectly, the induction of various stress response genes to counter oxidative damage. Furthermore, exposure of the pneumococcus to CSC has previously been reported by us and others to cause significant upregulation of the two-component regulatory system 11 (TCS11), which is involved in the induction of genes associated with biofilm formation6,13, the efflux of various chemical and heavy metal toxicants, and, importantly in the context of the current study, those involved in promoting vancomycin resistance10. However, the transcriptional mechanisms, including possible involvement of TCS11, which mediate induction of the pneumococcal erm(B) gene, as well as the possible involvement of its product, ribosomal methylase, in attenuating oxidative stress remain to be identified. Nevertheless, it is noteworthy that ribosomal RNA methylation has been linked to protection against environmental/oxidative stress in both Escherichia coli and Staphylococcus aureus 14,15.
CONCLUSIONS
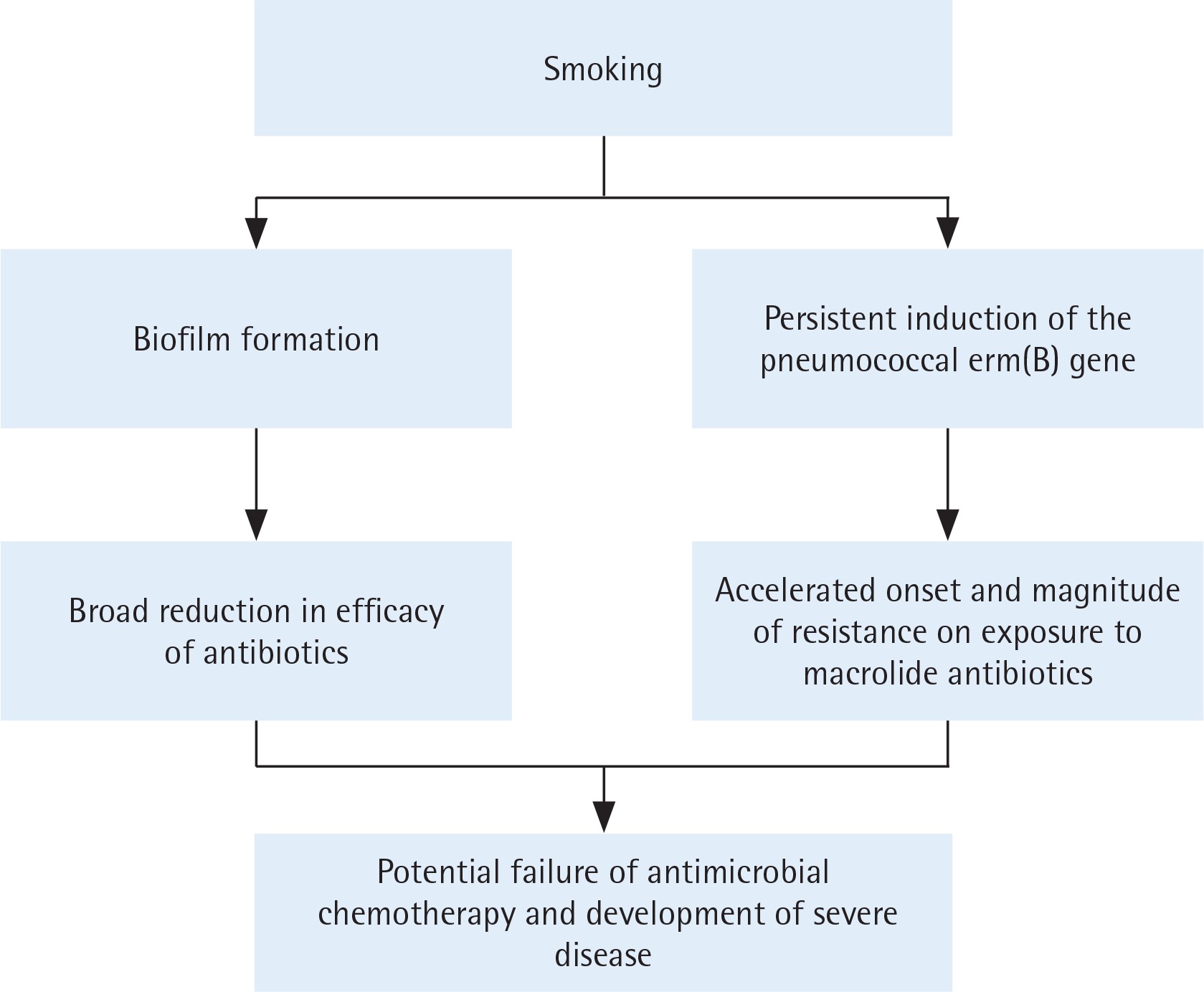
Based on the current and our previous studies, the mechanisms by which smoking may promote antibiotic resistance in the pneumococcus are summarized in Figure 2. Although the possible clinical significance remains to be established, these findings suggest that smoking may impede the efficacy of macrolidebased antimicrobial therapy by accelerating the onset and magnitude of erm(B)-mediated resistance, representing a novel pro-infective mechanism of smoking.